Prejavy ARG1-D sa často podobajú iným symptómom neurologických a neurometabolických porúch, ako sú iné poruchy močovinového cyklu (UCD), detská mozgová obrna (CP) alebo vrodená spastická paraplégia (HSP)5,6.
Diferenciálna diagnostika ARG1-D zahŕňa kombináciu klinických prejavov spojených s vysokými koncentráciami plazmatického arginínu4-7.
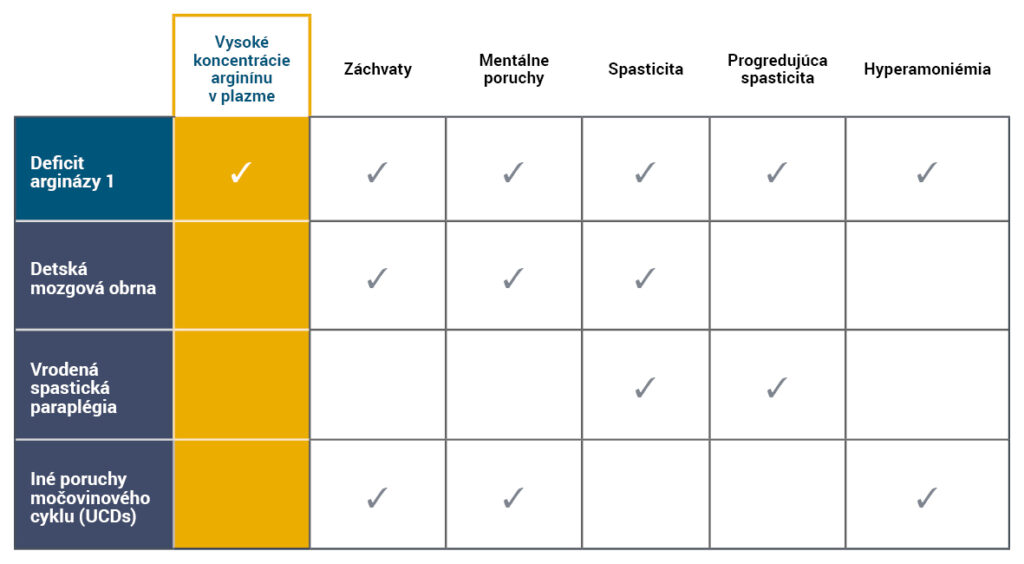
- Hyperamoniémia nie je typickým znakom ARG1-D a akútne epizódy hyperamoniémie sa vyskytujú len zriedkavo4,8.
Novorodenecký skríning má svoje limity, ktoré môžu viesť k falošne negatívnemu nálezu z mnohých dôvodov ako:
- Stanovenie hraničných koncentrácií arginínu pri skríningu je problematické, pretože prenos metabolitov, ako je arginín, z matky na dieťa môže ovplyvniť testovanie9,10.
- Skríningové algoritmy a hraničné koncentrácie arginínu sa líšia9.
- ARG1-D nie je súčasťou skríningových panelov novorodencov vo väčšine európskych krajín11.
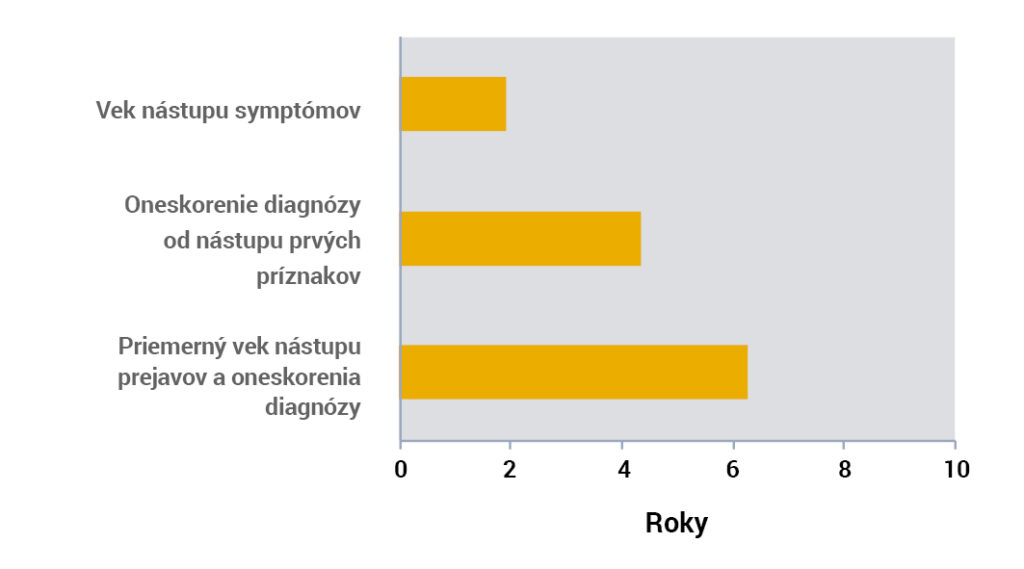
Oneskorenie diagnózy spojené s neskorým nástupom symptómov vedie k počiatočnej intervencii až vo veku ~ 6 rokov1.
ARG1-D môže byť zachytená v rámci analýzy hladín aminokyselín v krvi12,13.
Pred stanovením diagnózy je dôležité správne vyhodnotiť kompletnú zdravotnú, stravovaciu, rodinnú a sociálnu anamnézu a vykonať dôkladné fyzické vyšetrenie.
Overte zvýšenú koncentráciu arginínu, ktorá je typickým prejavom argininémie3,12,13.


Ak je koncentrácia arginínu v plazme zvýšená, môžeme diagnózu argininémie potvrdiť genetickým testom†.
†V dôsledku genetickej heterogenity genotypov ARG1 neboli identifikované všetky mutácie spôsobujúce ARG1-D.
Referencie:
1. Huemer M, et al. J Inherit Metab Dis. 2016;39:331-340. 2. Edwards RL, et al. J Inherit Metab Dis. 2009;32:S197-S200. 3. De Deyn PP, et al. Hyperargininemia: a treatable inborn error of metabolism. In: Guanidino Compounds in Biology and Medicine. London, UK: John Libbey Company Ltd; 1997:53-69. 4. Burrage LC, et al. Hum Mol Genet. 2015;24:6417-6427. 5. Carvalho DR, et al. Pediatr Neurol. 2012;46:369-374. 6. Prasad A, et al. J Child Neurol. 1997;12:301-309. 7. Crombez EA, Cederbaum SD. Mol Genet Metab. 2005;84:243-251. 8. Scaglia F, Lee B. Am J Med Genet C Semin Med Genet. 2006;142C:113-120. 9. Therrell BL, et al. Mol Genet Metab. 2017;121:308–313. 10. Pitt JJ. Clin Biochem Rev. 2010;31:57-68. 11. Loeber JG, Platis D, Zetterström RH et al. Int J Neonatal Screening. 2021;7:15. 12. Sun A, et al. Arginase deficiency. In: Adam MP, et al, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2020. 13. Ah Mew N, et al. Urea Cycle Disorders Overview. 2003. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1217/. Accessed November 26, 2021. 14. Cai X, et al. Medicine (Baltimore). 2018;97:e9880. 15. Bélanger SA, et al. Paediatr Child Health. 2018;23:403-410. 16. Lüneburg N, et al. J Nutr. 2011;141:2186-2190.